
La firma de tres genes revela cambios en el microambiente inmunológico del tumor en la progresión de NAFLD a HCC
Detección de genes pronósticos relacionados desde NAFLD/NASH hasta HCC
Para explorar los objetivos clave que regulan la transformación maligna de las lesiones hepáticas benignas, intentamos encontrar genes diferenciales (DEG) comunes en el desarrollo de lesiones hepáticas benignas y CHC. Utilizamos el paquete R “limma” para seleccionar 7668 grados en HCC de la base de datos TCGA, 458 y 8902 grados en NAFLD de GSE89632 y GSE126848, y 2804 y 6396 grados en NASH de GSE17470 y GSE126848, respectivamente (cambio logarítmico > 1, s<0,05) (Figura 1A). Se identificaron trece DEG como genes clave que regulan la transformación maligna de NAFLD/NASH (Figura 1B-D). Luego utilizamos datos de TCGA HCC para realizar análisis de regresión COX univariados y multivariados, e identificamos TEAD4, SOCS2 y CIT como los genes clave asociados con la transformación maligna de NAFLD/NASH y el pronóstico de CHC (Figura 1E,F). El análisis de correlación clínica mostró que la expresión de TEAD4 se asoció con un alto estadio T, estadio y grado de los tumores de CHC, la expresión de SOCS2 se asoció negativamente con metástasis en los ganglios linfáticos y el estadio T de los tumores de CHC, y CIT se asoció positivamente con el estadio T y el estadio de los tumores de CHC. . (Figura 1G-M). Entre ellos, TEAD4 (HR = 1,074) y CIT (HR = 1,595) son factores de riesgo para predecir el CHC, y su alta expresión conduce a un mal pronóstico del CHC, mientras que SOCS2 (HR = 0,791) es un factor protector, y su factor protector . La alta expresión mejora el pronóstico de los pacientes con CHC (Figura 1N-P).
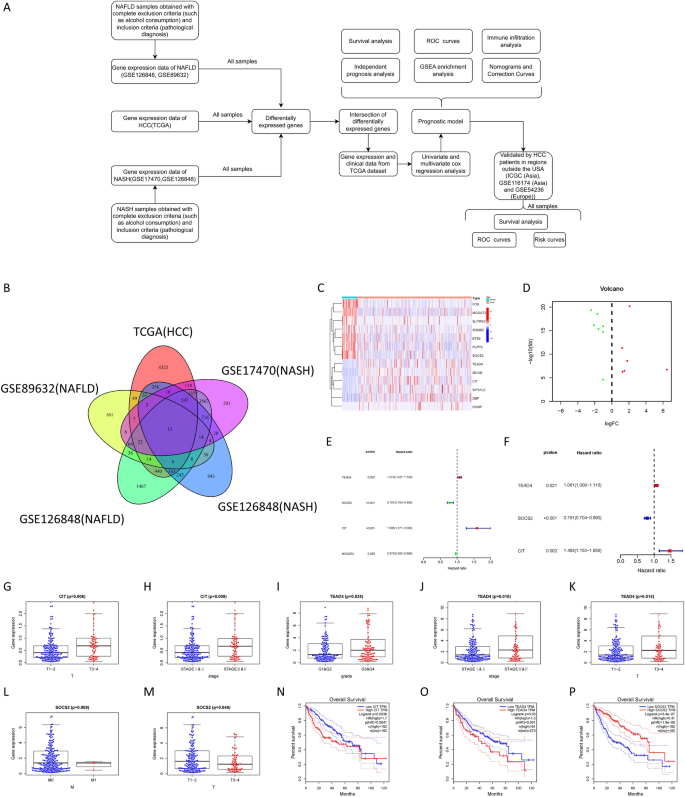
Detección de genes pronósticos relacionados desde NAFLD/NASH hasta HCC. (a) Diagrama de flujo del estudio. (B) Diagrama de Venn que muestra los genes diferenciales compartidos en NAFLD, NASH y HCC. (C, Dr) Mapa de calor (usando R versión 4.2.1: https://www.r-project.org/) y mapa de volcanes que muestra genes diferenciales en TCGA. E – F Análisis de regresión COX univariado y multivariado en TCGA. Análisis de correlación clínica de GM basado en la expresión génica. Análisis de la supervivencia de NP basado en la expresión génica.
Construyendo un modelo de pronóstico
A continuación, construimos un modelo de pronóstico de riesgo de CHC a través de TEAD4, SOCS2 y CIT, y puntuación de riesgo = (0,05957332 * expresión TEAD4 – 0,233885624 * expresión SOCS2 + 0,38039386 * expresión CIT). El análisis de regresión de Cox univariado utilizando datos de TCGA mostró que la puntuación de riesgo era un factor de riesgo para el cáncer de hígado (HR = 1,519), y el análisis de regresión de COX multivariado mostró que la puntuación de riesgo era un factor de pronóstico independiente para el cáncer de hígado (HR = 1,436) (Figura 2a, b). . Además, los pacientes con CHC con una puntuación de riesgo más alta mostraron un estadio T y un estadio más altos (Figura 2C,D). El análisis de supervivencia mostró que los pacientes con CHC con una puntuación de alto riesgo tenían un peor pronóstico en comparación con los pacientes con CHC con una puntuación de bajo riesgo (Figura 2E). Las curvas ROC de 1, 2 y 3 años (áreas bajo la curva ROC = 0,757, 0,751, 0,704) mostraron que nuestro modelo de pronóstico era confiable (Figura 2F). La Figura 2G-I muestra la expresión de TEAD4, SOCS2 y CIT en muestras de CHC y el estado de supervivencia de los pacientes con CHC según la puntuación de riesgo.
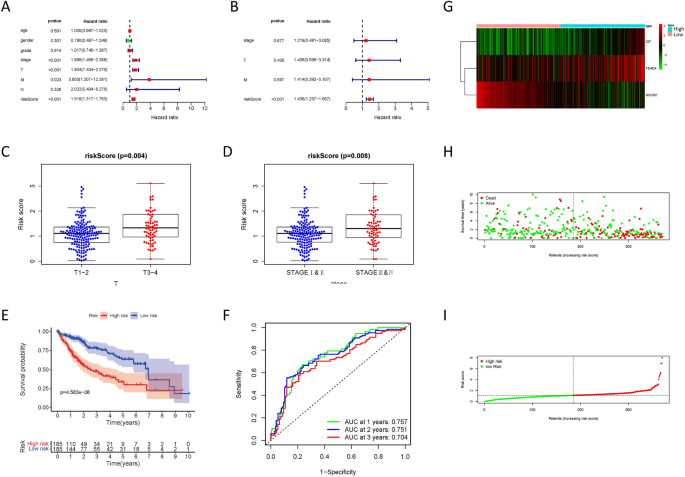
Construyendo un modelo de pronóstico. (a, B) Análisis de regresión COX univariado y multivariado basado en factores clínicos. (C, Dr) Análisis de correlación clínica basado en la puntuación de riesgo. (h, F) Curva de supervivencia y curva ROC según la puntuación de riesgo. (gramo–I) Expresión genética y estado de supervivencia según la puntuación de riesgo.
Validación del modelo de pronóstico.
Luego validamos nuestro modelo de pronóstico con conjuntos de datos externos (ICGC, GSE116174, GSE54236). Nuestros resultados mostraron que las puntuaciones de riesgo más altas se asociaron con menos especímenes supervivientes y más especímenes muertos (Figura 3A-I). El pronóstico de las muestras de bajo riesgo fue significativamente mejor que el de los grupos de alto riesgo (Figura 3J-L). En el conjunto de datos del ICGC, las áreas bajo la curva ROC para 1, 2 y 3 años son 0,656, 0,656 y 0,659, respectivamente; En el conjunto de datos GSE116174, son 0,604, 0,591 y 0,567; En el conjunto de datos GSE54236, son 0,791, 0,670 y 0,798, respectivamente (Figura 3M-O). Estos datos indican que nuestro modelo tiene un excelente valor pronóstico para los pacientes con CHC.
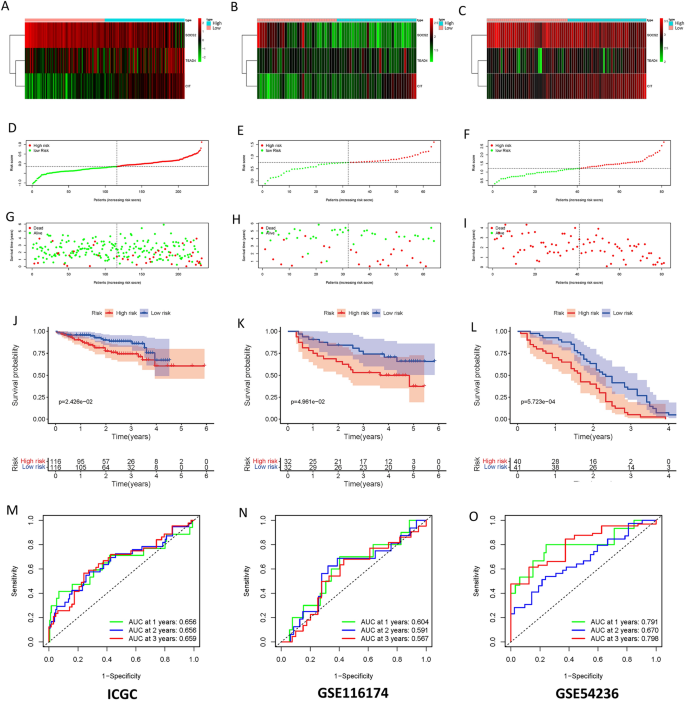
Validación del modelo de pronóstico. Mapa de calor de expresión genética (usando R versión 4.2.1): https://www.r-project.org/(para muestras de HCC del ICGC)a), GSE116174 (B) y GSE54236 (C). (Dr–F) Las calificaciones de riesgo para las muestras de CHC se distribuyen en los tres conjuntos de datos. (gramo–I) La relación entre la puntuación de riesgo, el estado de la muestra de CHC y el tiempo de supervivencia en los tres conjuntos de datos mencionados anteriormente. (C–a) muestras de las categorías de alto y bajo riesgo en las curvas de supervivencia de Kaplan-Meier para los tres conjuntos de datos mencionados anteriormente. (METRO–Ey) Curva ROC para puntuación de riesgo para muestras con cáncer de hígado.
Construcción y validación de nomogramas.
Para comodidad de los médicos, cuantificamos el modelo de pronóstico y combinamos otros factores de pronóstico independientes para desarrollar el nomograma. En la Figura 4A se muestran gráficos de OS a 1, 3 y 5 años. El índice de concordancia (índice C) del nomograma fue de 0,7 (se = 0,033), lo que indica un poder predictivo confiable del nomograma hasta cierto punto. Además, las curvas de calibración a 1, 3 y 5 años mostraron una alta precisión (Figura 4B-D).
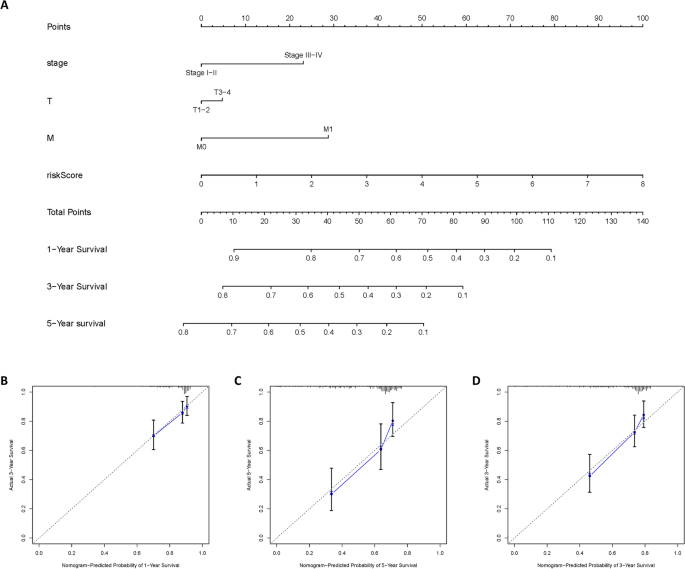
Construcción y validación de nomogramas. (a) Gráfico que muestra puntuaciones de supervivencia para factores clínicos. (B–Dr) Curvas de corrección que muestran precisión predictiva para la supervivencia a 1, 3 y 5 años.
Las características del entorno inmune del tumor y la infiltración inmune están significativamente asociadas con el grado de riesgo.
Dado que las anomalías inmunitarias son el mecanismo principal para la progresión de NAFLD/NASH a CHC, realizamos análisis relacionados con el sistema inmunológico basados en la puntuación de riesgo de las muestras de CHC. Los mapas de calor muestran la distribución de las células inmunitarias, la función inmunitaria y la puntuación inmunitaria en los grupos de alto y bajo riesgo (Figura 5A). En comparación con el grupo de bajo riesgo, el grupo de alto riesgo tuvo estimaciones de ImmuneScore más bajas, pero aumentó la pureza del tumor, mientras que ImmuneScore no fue significativamente diferente (Figura 5B-E). Mediante el análisis ssGSEA, los niveles de MSC infiltrantes, macrófagos y células Th2 aumentaron en el grupo de alto riesgo, mientras que los niveles de mastocitos infiltrantes, células asesinas naturales y células T auxiliares disminuyeron. En cuanto a las vías inmunes, se activó MCH I en el grupo de riesgo, IFN I y II, y se inhibió la actividad citolítica (Figura 5F). Mediante análisis de correlación, descubrimos que existía una asociación significativa entre los marcadores de riesgo y las células B infiltrantes (r = 0,205), células dendríticas (r = 0,321), células T CD4+ (r = 0,111) y células T CD8+ (r = 0,258). ), macrófagos (r = 0,332), neutrófilos (r = 0,256) (Figura 5G-L). Estos resultados indican que las respuestas inmunitarias, la infiltración inmunitaria y las firmas TIME están asociadas significativamente con las puntuaciones de riesgo en el CHC.
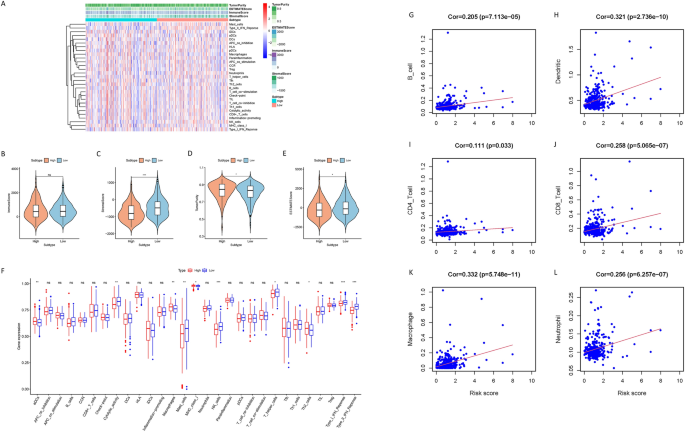
Las características del entorno inmune del tumor y la infiltración inmune están significativamente asociadas con el grado de riesgo. (a) Mapa de calor (usando R versión 4.2.1: https://www.r-project.org/) Distribución del grado de riesgo y distintos grados de inmunidad. (B–h) Comparación de la puntuación de grado, puntuación estromal, puntuación inmunológica y pureza del tumor entre grupos de alto y bajo riesgo. (F) La relación entre la función inmune, la infiltración de células inmunes y la puntuación de riesgo en el grupo TCGA. (gramo–a) La relación entre esta firma y CD4+ Células T, células B, CD8+ Células T, macrófagos, células dendríticas y neutrófilos.
El grado de riesgo está asociado con múltiples vías.
Para descubrir el mecanismo más profundo del mal pronóstico de los pacientes en el grupo de alto riesgo, realizamos un análisis de enriquecimiento GSEA por puntuación de riesgo y los resultados mostraron que KEGG_DNA_REPLICATION, KEGG_BASE_EXCISION_REPAIR, KEGG_RIBOSOME, KEGG_MISMATCH_REPAIR, KEGG_SPLICEOSOME, KEGG_HOMOLOGOUS_RECOMBINATION y otras vías se activaron significativamente. en pacientes del grupo de alto riesgo (Figura 6A-F). En el grupo de alto riesgo, múltiples vías metabólicas como KEGG_PRIMARY_BILE_ACID_BIOSYNTHESIS, KEGG_COMPLEMENT_AND_COAGULATION_CASCADES, KEGG_VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION, KEGG_FATTY_ACID_METABOLISM, KEGG_PROPANOATE_METABOLISM, KEGG_TRYPTOPHAN_TA fueron inhibidas significativamente ( Figura 6G-L).
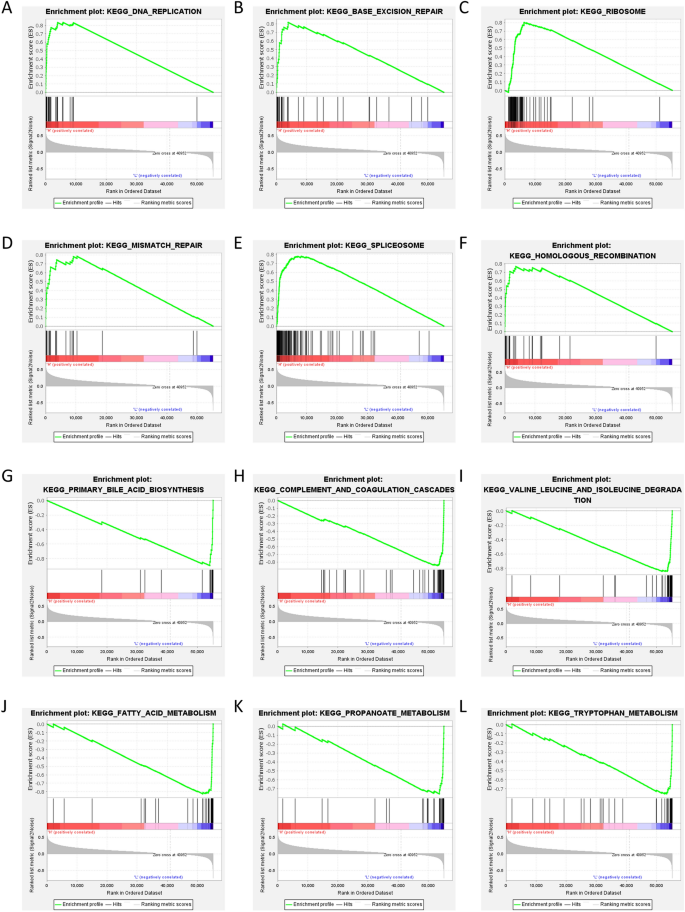
El grado de riesgo está asociado con múltiples vías. (a–F(El análisis GSEA revela 6 vías en la base de datos KEGG)www.kegg.jp/kegg/kegg1.html)31,32,33 Es activado por el grupo en riesgo en TCGA. (gramo–a) El análisis GSEA revela 6 vías en la base de datos KEGG (www.kegg.jp/kegg/kegg1.html)31,32,33Suprimido por el grupo en riesgo en la TCGA.
Patrones de expresión de genes clave para la transformación maligna de lesiones hepáticas benignas en NAFLD, NASH y HCC
Luego analizamos los patrones de expresión de TEAD4, SOCS2 y CIT en NAFLD, NASH y HCC. Nuestros resultados mostraron que la expresión de TEAD4 disminuyó en NAFLD y NASH, pero aumentó en HCC (Figura 7A-E). La expresión de SOCS2 disminuyó significativamente en NAFLD, NASH y HCC, lo que indica que la baja expresión persistente de SOCS2 condujo a una progresión maligna de lesiones hepáticas benignas (Figura 7G-K). La expresión de CIT disminuyó en dos conjuntos de datos de NAFLD (GSE89632 y GSE126848) y aumentó en HCC (Figura 7M-O). Sin embargo, CIT mostró cambios opuestos en los dos conjuntos de datos NASH (GSE17470 y GSE126848) (Figura 7P,Q). Debido a que NASH puede progresar a fibrosis esteatohepática, planteamos la hipótesis de que CIT disminuye en las primeras etapas de NAFLD y NASH, pero su expresión anormalmente alta podría mediar en la fibrosis de NASH. El análisis de la base de datos GSE89632 que contiene datos de estadificación de la fibrosis para NASH mostró que la expresión de CIT fue significativamente mayor en pacientes con cirrosis avanzada con NASH que en pacientes tempranos, lo que también es consistente con la mayor expresión de CIT en HCC (Figura 7R). Sin embargo, no hubo diferencias significativas en los niveles de expresión de TEAD4 y SOCS2 (Figura 7F,L). Esto sugiere que la alta expresión de CIT podría mediar el desarrollo de fibrosis NASH, lo que lleva a la aparición y progresión de HCC, y TEAD4 y SOCS2 pueden influir en el HCC a través de vías distintas a la fibrosis hepática.
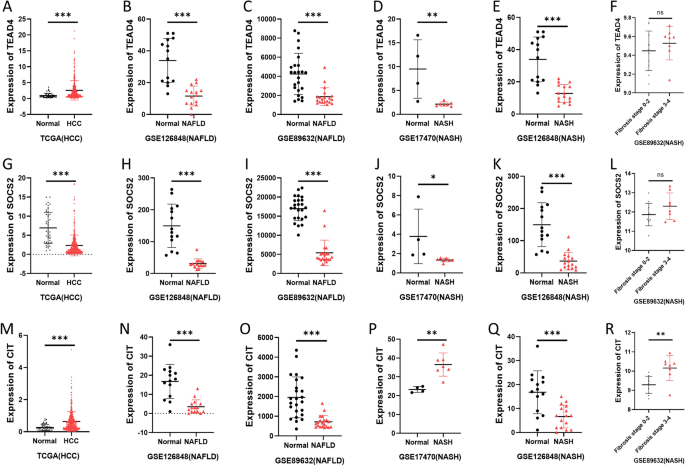
Patrones de expresión de genes clave para la transformación maligna de lesiones hepáticas benignas en NAFLD, NASH y HCC. TED4 (a–F), SOCS2 (gramo–a), y tecnología de la información (METRO–R) Niveles de expresión genética en conjuntos de datos NAFLD, NASH y HCC.
CIT se expresa altamente en muestras clínicas de CHC y se asocia con un mal pronóstico
Aún no existe ningún informe sobre las TIC en HCC. El análisis de supervivencia basado en la base de datos TCGA mostró que la tasa de supervivencia y la tasa de supervivencia libre de enfermedad de los pacientes con CHC con alta expresión de CIT fueron significativamente más bajas que las del grupo de baja expresión de CIT (Figura 8A, B). Los datos de HPA también mostraron una alta expresión proteica de CIT en tejidos de HCC (Figura 8C). A continuación, realizamos una detección de qPCR en 40 casos de tejidos de HCC del Hospital Oncológico de la Universidad Médica de Guangxi y sus tejidos cancerosos emparejados, lo que mostró que CIT se expresaba altamente en tejidos de HCC, lo que también fue confirmado por los resultados de la transferencia Western (Figura 1 a). . 8d,e). La alta expresión de CIT se asoció significativamente con un mal pronóstico de los pacientes con CHC (Figura 8F). Luego encontramos que CIT se expresaba altamente en todas las líneas celulares de HCC (Figura 8G, H). Construimos un modelo de sobreexpresión utilizando las líneas celulares SMCC-7721 y Hep-3B con expresión mínima (Figura 8I, J). El ensayo de proliferación celular y el ensayo CCK8 mostraron que CIT mejoró significativamente la capacidad de proliferación de las células HCC (Figura 8K, L).
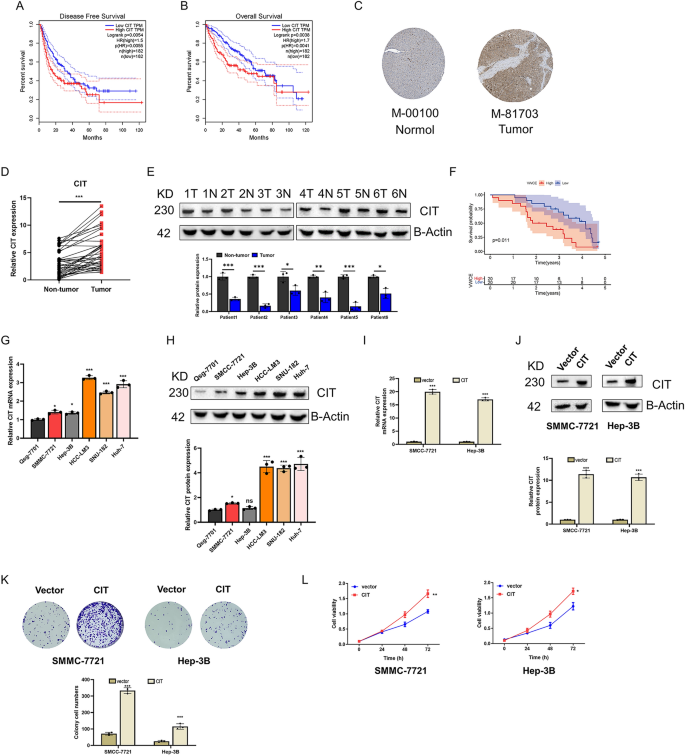
CIT se expresa altamente en muestras clínicas de CHC y se asocia con un mal pronóstico. (a) Curvas de supervivencia para pacientes con CHC de la base de datos TCGA. (B) Curvas de supervivencia libre de enfermedad para pacientes con CHC de la base de datos TCGA. (C) Resultados de IHC de la base de datos HPA que muestran la expresión de proteínas de CIT en tejidos de HCC. ARN (Dr) y proteína (h) Expresión de CIT en 40 HCC y tejidos tumorales del Hospital Oncológico de la Universidad Médica de Guangxi. (F) Curvas de supervivencia de 40 pacientes con cáncer de hígado del Hospital Oncológico de la Universidad Médica de Guangxi agrupados según la expresión de CIT. (gramo, h) Niveles de proteína y ARNm de CIT en líneas celulares de carcinoma hepatocelular. (I, C) Crear un modelo CIT de sobreexpresión. (k, a) Los ensayos de proliferación celular y CCK8 demuestran la capacidad proliferativa de las células HCC.

“Defensor de la Web. Geek de la comida galardonado. Incapaz de escribir con guantes de boxeo puestos. Apasionado jugador”.